 A couple of weeks ago Bryan gave you an overview on our sample database, but how do we get from those samples to creating a DNA test for breeders to use? Well, it’s a long process and this week we are going to explain how we use whole-genome sequencing (WGS) to find plausible candidate variants which are then taken into our lab for validation.
A couple of weeks ago Bryan gave you an overview on our sample database, but how do we get from those samples to creating a DNA test for breeders to use? Well, it’s a long process and this week we are going to explain how we use whole-genome sequencing (WGS) to find plausible candidate variants which are then taken into our lab for validation.
What is whole-genome sequencing (WGS)?
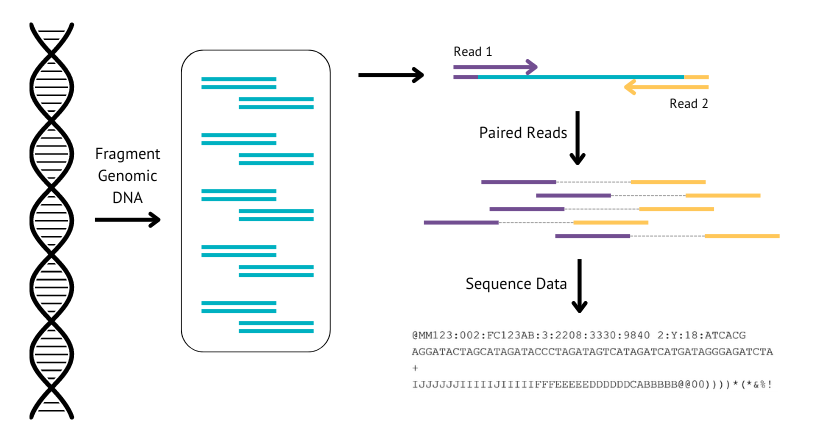 WGS, as the name suggests, involves sequencing the whole genome of the DNA extracted from the buccal swab. The canine genome is made up of 39 pairs of chromosomes (that’s 2.4 billion letters of DNA). Even with technology advances today, it is still very hard to sequence a whole chromosome in one go, so we use a process called short paired-end sequencing. This process involves breaking the DNA up into short bits of sequence (about 50o letters), which are then sequenced (“read”) from both ends. On average, each position in the genome is sequenced 30 times (called 30X coverage) to help reduce errors and get more accurate information. The actual sequencing is out-sourced and we receive text files back of all the short reads, called FASTQ files. The canine genome itself is around 2.4Gb in size , and at approx 30X coverage, we get around 70Gb of data back for every sample, which is the equivalent to 70,000 high quality photos, or 2 days of video!
WGS, as the name suggests, involves sequencing the whole genome of the DNA extracted from the buccal swab. The canine genome is made up of 39 pairs of chromosomes (that’s 2.4 billion letters of DNA). Even with technology advances today, it is still very hard to sequence a whole chromosome in one go, so we use a process called short paired-end sequencing. This process involves breaking the DNA up into short bits of sequence (about 50o letters), which are then sequenced (“read”) from both ends. On average, each position in the genome is sequenced 30 times (called 30X coverage) to help reduce errors and get more accurate information. The actual sequencing is out-sourced and we receive text files back of all the short reads, called FASTQ files. The canine genome itself is around 2.4Gb in size , and at approx 30X coverage, we get around 70Gb of data back for every sample, which is the equivalent to 70,000 high quality photos, or 2 days of video!
What happens next?
Once we have the paired-end read data back from the sequencing centre, it’s over to our Bioinformatician to put the jigsaw pieces back together, by aligning the reads to the Canis familiaris (Dog) reference genome, and then interrogating the data to find positions where the sample is different to the reference genome (variants). These processes are done on a high-performance compute cluster (HPC) with various scripts and programs. In total it takes around 24hours, per sample, to run, and at the end, we have data (around another 100Gb per sample) that we can visually, and computationally, inspect. Obviously, all dogs are different to each other at a genomic level and, on average, we will find 6 million variants in each sample… so how do we find the causative variant(s)?
Our databank of WGS samples
Comparing one single sample to the reference genome results in many, many variants. A lot of those variants are what make the breed the way it is, and different to a German Shepherd Dog (the current reference genome). Starting back in 2016 with the Give a Dog a Genome project, we have built a massive databank of WGS sequences which we use to filter those ~6M variants.
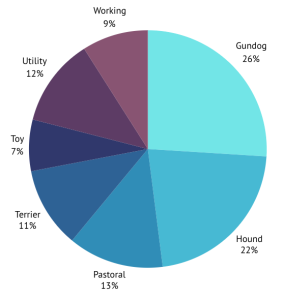
Over the last eight years the CGC has sequenced the genomes of 401 dogs, as well as downloading 61 extra genomes from publicly available databases, creating a databank of >450 genome sequences from 124 different breeds of dog, representing 21 clades and all seven UK breed groups. Just think how much storage space this takes up!! 462 samples at about 150Gb each… ~70TB!

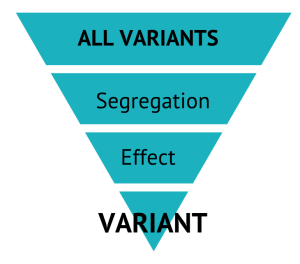 Comparing our “case” sample(s) against this whole databank of “control” samples, allows us to find genomic level differences between the two pools. We also look at the predicted effect that the variant would have at a biological level – the higher the effect prediction, the more likely the variant is to cause damage.
Comparing our “case” sample(s) against this whole databank of “control” samples, allows us to find genomic level differences between the two pools. We also look at the predicted effect that the variant would have at a biological level – the higher the effect prediction, the more likely the variant is to cause damage.
At this point we hope that we only have a single variant left from the original 6M… but more often than not, we have a handful. Returning to our sample database we use a random selection of breed-matched samples to validate the correct variant.
Comments are closed